El primer gen que se identificó como responsable de la Enfermedad de Stargardt, fue el gen ABCA4.
El gen ABCA4 presenta un gran tamaño y en él se pueden producir gran variedad de modificciones, hecho que se traduce en una amplia variabilidad clínica. En este sentido es necesario destacar que las nuevas técnicas de secuenciación masiva de genes se han convertido en instrumentos especialmente útiles para el diagnóstico de la enfermedad de Stargardt.
EL GEN ABCA4

En el año 1909, el Dr. Karl Bruno Stargardt, oftalmólogo alemán, describió una patología, cuyos afectados presentaban una atrofia macular.
Hoy, sabemos que la enfermedad de Stargardt, es una patología poco frecuente de origen genético, que se engloba dentro las Distrofias Hereditarias de la Retina y que es considerada la distrofia macular juvenil más común.
Los primeros síntomas pueden hacerse patentes en la primera o segunda década de vida, aunque en algunos casos aparecen en los primeros años de la vida adulta o incluso con posterioridad) y van variando con el paso del tiempo. Habitualmente debutan con reducción de la visión central (la visión más fina, la que nos aporta detalles) que puede progresar hasta llegar a lo que se considera ceguera legal (Agudeza Visual igual o inferior a 0,1/0,1), fotofobia, dificultad para adaptarse a condiciones de poca luminosidad, alteración en la percepción de los colores y fotofobia. La visión periférica suele mantenerse conservada.
 Pérdida de visión central Fotofobia
Pérdida de visión central Fotofobia
En 1997 se identificó por vez primera el gen responsable de esta enfermedad (ABCR). Concretamente “ATP-binding Cassette transporter gene, subfamilia A, miembro 4” de ahí su denominación como ABCA4.

Si observamos la fecha (1997), nos daremos cuenta de que el conocimiento sobre la implicación genética en esta patología es muy reciente. En biología y en ciencias un periodo de 20 años es un “suspiro”.
La aparición, en los últimos años, de técnicas de secuenciación masiva de genes junto con técnicas bioinformáticas para la interpretación de resultados, ha permitido grandes avances en el diagnóstico genético y en el descubrimiento de nuevas mutaciones.
“En los últimos años el avance de la secuenciación masiva (NGS) supone una valiosa herramienta para el estudio genético de estos pacientes porque permite secuenciar en poco tiempo y por un coste reducido desde un solo gen hasta el genoma completo en un solo ensayo, siendo de alta aplicabilidad al diagnóstico de las distrofias maculares”
«La secuenciación masiva (NGS) como método diagnóstico en la enfermedad de Stargardt». B. Jimenez-Rolando, S. Noval, I. Rosa-Perez, E. Mata Diaza, A. del Pozo, C. Ibañez, J.C. Silla , V.E.F. Montaño, R. Martin-Arenas y E. Vallespin. Arch Soc Esp Oftalmol 2018; 93:119-25

Secuenciador para ADN. Medicalexpo.es

ABCA4 se trata de un gen de gran tamaño, que se encuentra en el brazo corto del cromosoma 1 y que codifica para una proteína de los fotorreceptores que participa en el transporte de sustancias a través de membranas.
Un proceso que es fundamental para nuestro desarrollo y crecimiento es alimentarnos, es decir nutrirnos; lo mismo ocurre para el óptimo desarrollo de nuestras células.

El proceso de nutrición de nuestras células, absorción de nutrientes y eliminación de residuos se realiza a través de las membranas celulares. Estos procesos de tránsito a través de las membranas se encuentran regulados por proteínas codificadas por la información que tenemos en nuestros genes.
Cuando una mutación afecta a una proteína que regula el tránsito de sustancia a través de las membranas, puede ocurrir que la célula no se nutra de manera adecuada o que los residuos que han de ser eliminados no salgan de la célula, quedándose acumulada en ella.
En la enfermedad de Stargardt, las mutaciones en el gen ABCA4, provoca la acumulación de subproductos tóxicos de la Vitamina A, produciendo un deterioro gradual de los fotorreceptores (células responsables del proceso de fototransducción) y, por tanto, la pérdida de la visión.
LA VITAMINA A Y EL CICLO VISUAL
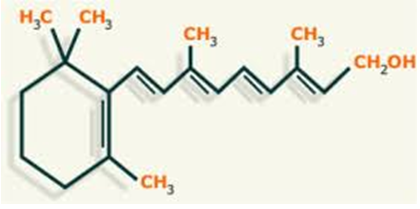
La vitamina A se obtiene en forma de retinilo de los alimentos de origen animal y en forma de betacaroteno en los de origen vegetal.
El retinilo, al llegar al intestino, es transformado en retinol, forma activa de la Vitamina A. El retinol es transportado hacia la retina, donde se transforma en retinal y se une a la proteína opsina de los fotoreceptores formando la rodopsina, el pigmento visual. Una vez utilizada, los metabolitos de la vitamina A son eliminados.
“Las mutaciones en el gen ABCA4 conlleva que las células fotorreceptoras de la retina, es decir, los bastones y los conos, de los individuos afectados no pueden llevar a cabo de forma eficaz su función en el proceso denominado ciclo visual. En concreto, el gen ABCA4 mutante no funciona en la parte del ciclo visual donde la vitamina A se transporta en ambas direcciones entre las células fotorreceptoras y una capa de células vecinas denominada epitelio pigmentario de la retina (EPR). En consecuencia, en el interior del EPR se produce un derivado tóxico de la vitamina A, llamado A2E, que se acumula en forma de unos depósitos de color amarillento claro llamados lipofucsina”.
GENETICA Y PATRONES DE HERENCIA
En este punto, vamos a describir de manera sencilla, dos de los diversos patrones de herencia, que se conocen.
- Herencia Autosómica Recesiva
- Herencia Autosómica Dominante
Empecemos por conocer de manera muy sencilla qué son genes y cromosomas.
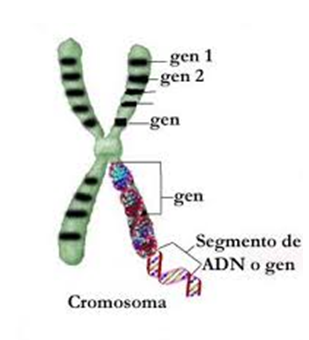
Un gen es básicamente información; ésta hace referencia a rasgos físicos visibles, como puede ser el color de nuestro pelo, ojos…, pero no es solo eso, la información contenida en los genes también indica a la célula qué funciones ha de realizar, qué sustancias debe fabricar para su correcto funcionamiento, etc.
Cuando la información contenida en los genes está alterada tendremos una mutación, como consecuencia, la información destinada a indicar a la célula qué funciones ha de realizar o qué productos ha de sintetizar no es correcta. Esto comporta que estos procesos no se produzcan o los que se producen no funcionen de forma adecuada.
Los genes, son fragmentos de ADN, que se encuentran en los cromosomas. Podemos definir los cromosomas como las estructuras que se encuentra en nuestra célula y en cuyo interior llevan la información genética.
Nuestras células contienen 23 pares de cromosomas: 22 autosomas y los cromosomas sexuales que son los que determinan nuestro sexo, XY para el hombre y XX para la mujer.
Teniendo esto en cuenta, la Herencia Autosómica hace referencia al hecho de que la información genética o gen que estamos estudiando, se encuentra en uno de los cromosomas que forma parte de esos 22 pares a los que denomínanos autosomas y que por lo tanto no está ligada al sexo.
Biológicamente todos necesitamos de un progenitor masculino (padre) y otro femenino (madre). Esto hace que la información genética que tenemos para cada carácter o característica que estudiemos esté duplicada. Así, la expresión de un carácter vendrá definido por la información que para ese carácter aporte nuestro padre y la que aporte nuestra madre. Cada una de estas copias se llaman alelos; un alelo lo aportará nuestra madre y el otro nuestro padre. Estos alelos pueden de iguales o diferentes.
En la población humana y animal hay unos determinados rasgos que son más comunes que otros, como ejemplo: al analizar el color de los ojos, el color marrón es más común que el color azul y em general un color oscuro de ojos es más común que un color claro, por ello decimos que el color marrón o el color oscuro es dominante sobre el color azul o el color claro de los ojos.
Podemos decir que el carácter “color de ojos marrón” es dominante y el carácter “color de ojos azul” es recesivo.
Indicamos que un patrón de herencia es dominante, cuando los dos alelos o al menos uno lleva información dominante para el carácter o característica en estudio. Hablamos de un patrón de herencia recesivo, cuando los dos alelos llevan información recesiva para el carácter o característica en estudio.
Usando, como ejemplo el color de ojos marrón y azul, tendremos:
- Si mis alelos, materno y paterno, llevan información para color de ojos marrón, lo normal es que yo tenga los ojos marrones.
- Si un alelo lleva información para color de ojos marrón y el otro azul, dado que el color de ojos marrón es dominante, yo tendré los ojos marrones.
- Si mis alelos, paterno y materno, llevan información para color de ojos azules, yo tendré los ojos azules. Para que se exprese el carácter recesivo, siempre, es necesario la presencia de esa información en los dos alelos (paterno y materno)
(Este ejemplo es una simplificación, que solo intenta explicar de forma muy sencilla dominancia y recesividad, sin duda la expresión génica es mucho mas compleja y sujeta a procesos que no se tratan es este artículo)
ENFERMEDAD DE STARGARDT: TIPOS Y MUTACIONES ASOCIADAS
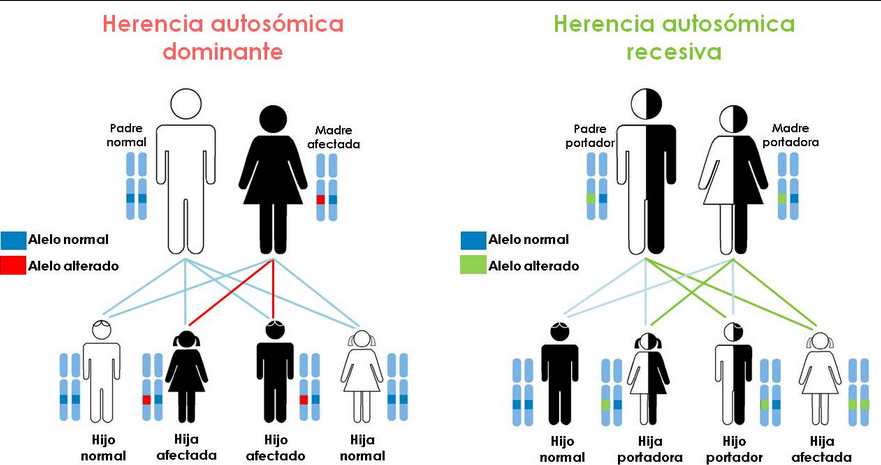
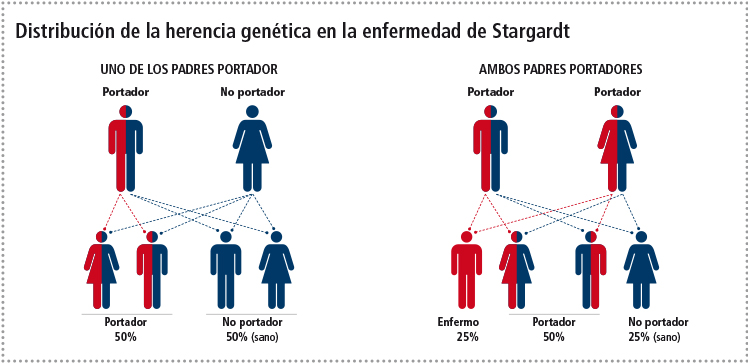
La enfermedad de Stargardt es la forma más frecuente de distrofia macular de inicio juvenil, fundamentalmente, debido a mutaciones genéticas según un patrón hereditario autosómico recesivo.
La denominación autosómica indica que la mutación se encuentra en un cromosoma diferente al que determina el sexo y al ser recesiva implica que la mutación ha de estar presente en el alelo que aporta el padre y en el que aporta la madre.
Si bien este patrón de herencia es el más común, lo cierto es que la enfermedad tiene una gran variabilidad clínica y genética.
La enfermedad de Stargardt es una enfermedad ocular progresiva que compromete la función de la parte central de la retina también conocida como la mácula. La retina es un tejido complejo que recubre la superficie interior de la parte posterior del ojo, y que es responsable de convertir la luz que recibimos del mundo exterior en una señal visual en nuestro cerebro. En la actualidad esta patología es incurable ya que no existe aprobada ninguna terapia.
La enfermedad de Stargardt está asociada a una considerable variabilidad clínica y genética, se han descrito mutaciones, en al menos tres genes, lo que ha permitido categorizar la enfermedad.
Enfermedad de Stargardt tipo 1 (STGD 1)
Es la forma más común de STGD. Está causada por mutaciones en el gen ABCA4. Como hemos visto previamente, se trata de un gen de gran tamaño, que se encuentra en el brazo corto del cromosoma 1 y que codifica para una proteína de los fotorreceptores que participa en el transporte de sustancias a través de membranas, concretamente “ATP-binding Cassette transporter gene, subfamilia A, miembro 4”.
Alrededor de dos tercios de las personas con STGD, tienen mutado este gen, habiéndose descrito mas de 600 mutaciones causantes de enfermedad en este gen.
El patrón de herencia, en STGD 1, es autosómico recesivo, lo que implica que las mutaciones no están ligadas al sexo y las personas afectadas tienen mutado tanto el alelo materno como el paterno. Normalmente los padres de los afectados son portadores sanos de la enfermedad, siendo posible encontrar uno o varios hermanos enfermos.
Los descendientes de los afectados serán portadores sanos de la enfermedad, siempre que la pareja de la persona afectada sea sana no portadora.
Si es importante destacar que, si bien la enfermedad de Stargardt tipo 1 se caracteriza por las mutaciones en el gen ABCA4, hasta la fecha se han descrito mutaciones en ABCA4 en diferentes enfermedades: Retinosis pigmentaria, Distrofia de conos y bastones, Enfermedad de Stargardt y Degeneración Macular Asociada a la Edad.
Enfermedad de Stargardt tipo 3 (STGD 3)


STGD 3, es una forma autosómica dominante de enfermedad de Stargardt. Se trata de una forma poco común, causada por el gen ELOVL4, que se encuentra en el brazo corto del cromosoma 6.
ELOVL4 se expresa en los segmentos internos de los fotorreceptores. El gen codifica para una proteína que se une a la membrana de los fotorreceptores y está implicada en la elongación de los ácidos grasos de cadena larga.
No se sabe como la actividad de la proteína defectuosa expresada por el gen ELOVL4, llega a acumular lipofuscina
Por tener un patrón de herencia dominante, es importante destacar, que para que se exprese la enfermedad solo es necesario tener mutado uno de los alelos, bien el aportado por la madre o el aportado por el padre y los descendientes de los afectados por este tipo de STGD tienen un 50% de posibilidades de ser afectados, suponiendo que la pareja del afectado sea sana.
Enfermedad Stargardt tipo 4 (STGD 4)


Es la forma más reciente de STGD en ser descrita.
La enfermedad es causada por una mutación en el gen PROM1. Este gen se encuentra localizado en el brazo corto del cromosoma 4 codificando para una proteína llamada Prominin 1 (PROM1, también conocido como CD133 y AC133). PROM1 participa en la formación y organización de los discos dentro de los fotorreceptores.
Los fotorreceptores, conos y bastones, en su estructura cuentan con una membrana que se va plegando formando unos elementos denominados discos que son fundamentales para la función visual. Los discos son el sitio del fotoreceptor donde se produce la transformación fotoquímica de la luz.
La Proteína PROM1 defectuosa no puede migrar hacia la zona de los fotorreceptores donde se forman los discos, por lo que la ausencia de la proteína lleva, en última instancia, al incorrecto funcionamiento de los discos y a los problemas visuales.
Recientemente se ha descubierto que las mutaciones en el gen PROM1 podrían transmitirse como un rasgo autosómico dominante y/o como un rasgo autosómico recesivo. La forma dominante autosómica se asocia más con el STGD4 y otras distrofias maculares, mientras que el rasgo recesivo está más relacionado con enfermedades como la distrofia de conos y bastones y retinosis.
Revisado estos tres tipos de enfermedad de Stargardt, parece que nos hemos dejado algo por el camino, pero no es así, hemos dejado para el final la enfermedad de Stargardt tipo 2, dado que es el tipo del que menos información se posee.
Enfermedad de Stargardt Tipo 2 (STGD 2)

En relación con este tipo de enfermedad hay poca información. En 1994 se encontró una familia con una variante de la enfermedad a la que se denominó enfermedad de Stargardt de tipo 2 (STGD2), y en la que el gen responsable se localizó en el brazo corto del cromosoma 13.
RESUMEN
La enfermedad de Stargardt, es una patología con una gran variabilidad clínica y genética. Se considera la distrofia juvenil mas común, pero puede ser diagnosticada en diferentes etapas de la vida: infancia, adolescencia, primeros años de la edad adulta o con posterioridad.
Los síntomas pueden empezar en la primera o segunda década de vida con una progresión que varía con el tiempo.
Es patente una reducción de la visión central, con perdida en la agudeza visual, que puede llevar a la ceguera legal, pero manteniendo la visión periférica.
Además de la pérdida progresiva de la visión central que causa visión borrosa, en ocasiones nos podemos encontrar con dificultad creciente para adaptarse a la oscuridad. La mayoría de las personas afectadas también tienen problemas de la visión del color. La fotofobia puede estar presente.
Es difícil predecir cuándo se manifestará la enfermedad y como de rápida será la progresión. La evolución de la enfermedad varia de persona a persona e incluso cuando hay mas de un miembro afectado en la misma familia, la evolución de la enfermedad es diferente entre ellos.
Desde un punto de vista genético, la forma más habitual de enfermedad de Stargardt, es la causada por mutaciones en el gen ABCA4, siendo el patrón de herencia más común el autosómico recesivo, en el que la persona afectada ha de tener alteradas las dos copias de este gen: la que aporta su padre y la que aporta su madre, siendo los padres sanos para esta enfermedad.
Las modernas técnicas de secuenciación masiva de genes nos aportan nueva información sobre el carácter genético de la enfermedad, encontrando nuevos genes implicados como ELOVL 4 y PROM 1, encontrándonos con casos en los que el patrón de herencia no es recesivo, sino dominante, lo que favorece la existencia de familias con muchos individuos afectados.
Estos nuevos genes codifican para proteínas diferentes a la que codifica ABCA 4, por lo que los acúmulos de lipofuscina pueden no ser la única vía que lleve a producir la sintomatología de esta enfermedad, pudiendo estar implicados fenómenos de elongación de ácidos grasos de cadena larga, la formación de los discos presentes en la estructura de conos y bastones.
Sin duda alguna, la mejora de las técnicas de diagnóstico genético, nos abrirá nuevas puertas al conocimiento de la Enfermedad de Stargardt y a posibles vías de tratamiento.
Referencias:
1.- Enfermedad de Stargardt. Barcelona Macula Fundation. https://barcelonamaculafound.org/es/patologias/enfermedad-de-stargardt/
2.- Fundus Flavimaculatus y Enfermedad de Stargardt. Dra. Rosa María Coco Martín. http://www.retinanavarra.org/images/documentos/stargardt04.pdf
3.- Programa Progstar. http://progstar.org/
4.- «La secuenciación masiva (NGS) como método diagnóstico en la enfermedad de Stargardt». B. Jiménez-Rolando, S. Noval, I. Rosa-Pérez, E. Mata Diaz, A. del Pozo, C. Ibáñez, J.C. Silla, V.E.F. Montaño, R. Martin-Arenas y E. Vallespín. Arch Soc Esp Oftalmol 2018; 93:119-25
5.- Stargardt, Las personas con enfermedad de Stargardt o distrofia de conos y bastones recesiva no deberían exceder la cantidad diaria recomendada de vitamina A. Traducido por José Martín Nieto. G Humana . 2008
6.- Preparando la nueva comunidad sobre la enfermedad de Stargardt. Rare commons. Sant Joan de Déu Barcelona Hospital. https://www.rarecommons.org/es/actualidad/preparando-nueva-comunidad-sobre-enfermedad-stargardt
7.- Vitamina A. Linus Pauling Institute. Oregon State University https://lpi.oregonstate.edu/es/mic/vitaminas/vitamina-A
Aquí os dejamos una pintura de una obra de Joaquín Sorolla, el pintor de la luz.
